
Published on May 20, 2024
Table of Contents
Overview

In this article, we are going to explore Slurm, a popular open-source high-performance computing (HPC1) workload manager, and discover what it is, why people use it, and how it differs from Kubernetes.
For context, when we talk about high-performance computing, we are primarily talking about computational tasks that require a lot of time on very powerful computer systems and are often designed, so that they can be highly parallelized, across resources within a super-computer or large cluster.
These sorts of jobs are very common in scientific research, computer simulation, artificial intelligence, and machine learning tasks.
What is Slurm?
To almost directly quote the Slurm repository README.
Slurm is an open-source cluster resource management and job scheduling system for Linux (primarily) that strives to be simple, scalable, portable, fault-tolerant, and interconnect agnostic. As a cluster resource manager, Slurm provides three key functions:
- First, it allocates exclusive and/or non-exclusive access to resources (compute nodes) to users for some duration of time so they can perform work.
- Second, it provides a framework for starting, executing, and monitoring work (normally a parallel job) on the set of allocated nodes.
- And finally, it arbitrates conflicting requests for resources by managing a queue of pending work.
In short, Slurm is a system that is primarily designed to manage batch processing workloads running in a shared computing cluster. Slurm can be configured to run any type of batch job but is often utilized on systems that support multiple users with parallel computing tasks to run.
According to SchedMD, the primary support and development company for the Slurm community, Slurm is being used on approximately 65% of the TOP500 supercomputers in the world.
A bit of history
In the mid-1990s, as Linux became more popular, there was an increased interest in making large-scale computer processing more accessible through the use of commodity hardware and projects like Beowulf.
In 2001, engineers at the Lawrence Livermore National Laboratory (LLNL2) in the United States, started looking into how they might migrate from proprietary HPC systems to open-source solutions. They determined that there weren’t any good resource managers available at the time, and this directly led to the creation of Slurm, which was initially released in 2002.
The name Slurm is a reference to the soda in Futurama.
Over the years, Slurm has evolved to support a wide variety of hardware configurations and in 2010, some initial scheduling logic was added to Slurm, enabling it to function as both a stand-alone resource manager and a scheduler3.
The Architecture
Like many similar systems, a Slurm setup consists of at least one controller node, and at least one compute node, which can be on the same physical systems.
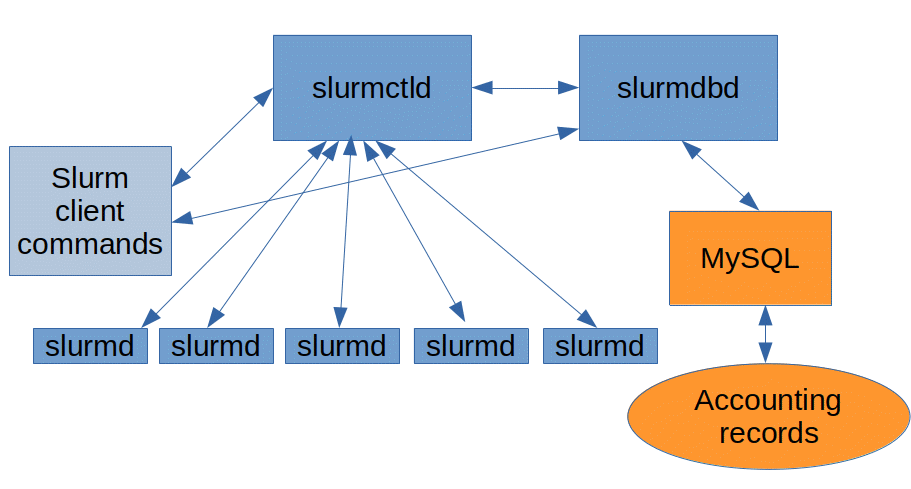
The Controller
Two daemons are responsible for managing the cluster. These two processes can be run on a single node or separated, depending on the performance and resiliency required by the administrator.
slurmctld
slurmctld is the primary management daemon for Slurm. It is responsible for all the daemons, resources, and jobs, within the cluster. Because it is a critical component in the cluster, it is often configured with a passive backup that can take over management if the primary daemon ever fails.
slurmdbd
slurmdbd provides an interface for the MySQL database that Slurm uses for stateful data, like job accounting records.
Compute Nodes
These represent the nodes whose resources are available to any jobs that might need to be scheduled in the cluster.
slurmd
slurmd is the daemon that runs on each compute node and is responsible for accepting work, launching tasks, and generally managing the workloads on each node.
Installation
SchedMD maintains the core documentation for Slurm and the current installation documentation can be found on their website.
Getting Slurm working is not particularly difficult, but fine-tuning it for your requirements can be a time-consuming task. Since performance is often a primary focus with these clusters and Slurm is designed to work with a very wide set of hardware and software libraries, it can take a lot of effort to fine-tune the installation, so that it utilizes the hardware to the best of its ability, and all the proper software is available to streamline the types of jobs that will be run on the cluster.
At a minimum clusters will require node name resolution, a MySQL instance, shared block storage, a synchronized time source, synchronized users, the Munge daemon for user authentication, a full set of custom-built Slurm executables, and various configuration files.
In addition to this, the nodes will need to contain the various hardware, programming languages, libraries, and other tools that the majority of jobs will require to run. This could include things like GPUs4 and their related toolchains (CUDA5/cuDNN6), MPI7 libraries, OCI8 runtimes for container support, and much more.
Slurm’s greatest asset and liability is how flexible it is. This makes it something that can be highly optimized for the requirements of the organization, while also making it susceptible to misconfiguration.
Workloads
To provide some context, let’s take a look at an example user job and workflow that utilizes some of the features of Slurm.
In the mpi-ping-pong.py code below, you will find a Python script that simulates a pseudo-game of Ping Pong/Table Tennis by making use of MPI to pass a message (“ball”) between as many processes as we have configured to participate in the parallel job.
Local Testing
To run this code, you simply need to have Python, OpenMPI, and mpi4py installed on your system.
mpi-ping-pong.py
#!/usr/bin/env python
# Based on examples from:
# https://pdc-support.github.io/introduction-to-mpi/aio/index.html
#
# But expanded to be more verbose and work in more use cases.
from mpi4py import MPI
size = MPI.COMM_WORLD.Get_size()
rank = MPI.COMM_WORLD.Get_rank()
name = MPI.Get_processor_name()
rrank = rank + 1
# Introduce ourselves
print("Hello, World! I am 🏓 Player (Rank) ", rank, ". Process ", rrank ," of ",size," on ",name, flush=True)
max_count = 300 * size
ball = 1
# Find the neighbor we receive the ball from and the neighbor we send the ball to.
if rank == 0:
if size == 1:
rn = 0
sn = 0
else:
rn = size - 1
sn = rank + 1
elif rank != 0 and rrank == size:
rn = rank - 1
sn = 0
else:
rn = rank - 1
sn = rank + 1
# Announce who our neighbors are
print("Player (Rank) ", rank, "'s receiving 🏓 neighbor is Player (Rank) {:d} and my sending 🏓 neighbor is Player (Rank) {:d}.".format(rn,sn), flush=True)
# Tracking the counter is not a great method here since we have messaging, but it works...
if rank == 0:
counter = 1
else:
counter = 0
# If we are Rank 0, we get to start the match.
if rank == 0:
# Rank 0 starts with the ball
req = MPI.COMM_WORLD.isend(ball, dest=sn, tag=0)
if size == 1:
print("Player {:d} started round {:d} by hitting the 🏓 ball towards the wall.".format(rank,counter), flush=True)
else:
print("Player {:d} sent the 🏓 ball to Player {:d}.".format(rank,sn), flush=True)
bored = False
last = False
# Now run a send and receive in a loop until someone gets bored
while not bored:
if counter <= max_count:
# Receive the ball
req = MPI.COMM_WORLD.irecv(source=rn, tag=0)
recv_message = req.wait()
if size == 1:
print("Player {:d} received the 🏓 ball that bounced off the wall.".format(rank), flush=True)
else:
print("Player {:d} received the 🏓 ball from Player {:d}.".format(rank,rn), flush=True)
# Increment the counter and send the ball
counter += 1
# We don't want to send a ball once we are done playing for the moment.
if counter <= max_count:
# Determine if we are the last player standing.
if (rrank == size) and (counter == max_count):
last = True
else:
req = MPI.COMM_WORLD.isend(ball, dest=sn, tag=0)
if size == 1:
print("Player {:d} started round {:d} by hitting the 🏓 ball towards the wall.".format(rank,counter), flush=True)
else:
if rank == 0:
print("Player {:d} started round {:d} by sending the 🏓 ball to Player {:d}.".format(rank,counter,sn), flush=True)
else:
print("Player {:d} sent the 🏓 ball to Player {:d}.".format(rank,sn), flush=True)
# Check if the player is bored
bored = (counter >= max_count)
# Announce that we are done for the day.
if last == True:
if size == 1:
print("🎉 Player {:d} won the game of 🏓 ping pong, since they were playing alone! 🎉".format(rank), flush=True)
else:
print("🎉 Player {:d} won the game of 🏓 ping pong, since everyone else quit! 🎉".format(rank), flush=True)
else:
print("🛑 Player {:d} is bored of 🏓 ping pong and is quitting! 🛑".format(rank), flush=True)
mpi-ping-pong.py is what many would refer to as an embarrassingly parallel workload because it can be scaled with no real effort at all.
If you run this Python application on your local system, you will see something like this:
$ python ./mpi-ping-pong.py
Hello, World! I am 🏓 Player (Rank) 0 . Process 1 of 1 on laptop.local
Player (Rank) 0 's receiving 🏓 neighbor is Player (Rank) 0 and my sending 🏓 neighbor is Player (Rank) 0.
Player 0 started round 1 by hitting the 🏓 ball towards the wall.
Player 0 received the 🏓 ball that bounced off the wall.
Player 0 started round 2 by hitting the 🏓 ball towards the wall.
Player 0 received the 🏓 ball that bounced off the wall.
…
Player 0 started round 299 by hitting the 🏓 ball towards the wall.
Player 0 received the 🏓 ball that bounced off the wall.
🎉 Player 0 won the game of 🏓 ping pong, since they were playing alone! 🎉
As you can see the application runs perfectly fine, and uses a single rank9/process (CPU10 core, in this case). Since we only have a single process participating in this game of Ping Pong, the program simply sends and receives messages to and from from itself.
However, assuming that we have at least two CPU cores, then we can run this application utilizing as many of them as we want, by leveraging the MPI command line tool mpirun.
To run 12 instances of our application, which will all communicate with each other in parallel across 12 cores on our system, we can do the following on our local system:
NOTE: These messages will be somewhat out of order. This is the nature of parallel computing, buffers, etc. To deal with this you would likely need to send all the messages back to single process for ordering and printing.
$ mpirun -np 12 ./mpi-ping-pong.py
Player (Rank) 0 's receiving 🏓 neighbor is Player (Rank) 11 and my sending 🏓 neighbor is Player (Rank) 1.
Hello, World! I am 🏓 Player (Rank) 0 . Process 1 of 12 on laptop.local
Player (Rank) 1 's receiving 🏓 neighbor is Player (Rank) 0 and my sending 🏓 neighbor is Player (Rank) 2.
Hello, World! I am 🏓 Player (Rank) 1 . Process 2 of 12 on laptop.local
Player (Rank) 2 's receiving 🏓 neighbor is Player (Rank) 1 and my sending 🏓 neighbor is Player (Rank) 3.
Hello, World! I am 🏓 Player (Rank) 2 . Process 3 of 12 on laptop.local
…
🛑 Player 0 is bored of 🏓 ping pong and is quitting! 🛑
🛑 Player 1 is bored of 🏓 ping pong and is quitting! 🛑
🛑 Player 2 is bored of 🏓 ping pong and is quitting! 🛑
…
🎉 Player 11 won the game of 🏓 ping pong, since everyone else quit! 🎉
Massive Scale with Slurm
Now, imagine that we want to run this across hundreds of CPUs. Slurm makes this trivial as long as you have enough systems in your cluster to support this work.
In Slurm, you can define a batch job with a shell script (e.g. /shared_storage/mpi-ping-pong.slurm) that looks something like this:
#!/bin/bash
#SBATCH -J mpi-ping-pong # Name the job
#SBATCH -o /shared_storage/mpi-ping-pong-%j.out # The standard output file
#SBATCH -e /shared_storage/mpi-ping-pong-%j.err # The standard error file
#SBATCH -t 0-2:00:00 # Run for a max time of 0d, 2h, 0m, 0s
#SBATCH --nodes=3 # Request 3 nodes
#SBATCH --ntasks-per-node=1 # Request 1 cores or task per node
echo "Date start = $(date)"
echo "Initiating Host = $(hostname)"
echo "Working Directory = $(pwd)"
echo ""
echo "Number of Nodes Allocated = ${SLURM_JOB_NUM_NODES}"
echo "Number of Tasks Allocated = ${SLURM_NTASKS}"
echo ""
mpirun python /shared_storage/mpi-ping-pong.py
RETURN=${?}
echo ""
echo "Exit code = ${RETURN}"
echo "Date end = $(date)"
echo ""
Each #SBATCH line defines an sbatch command line argument to be used for the job. In this case, we are going to request that the workload run on three nodes and that it will only use a single core on each node.
Assuming that all the compute nodes in our cluster are configured correctly and our Python script exists on the shared storage at /shared_storage/mpi-ping-pong.py then we can submit this job to the cluster, by running:
$ sbatch /shared_storage/mpi-ping-pong.slurm
Submitted batch job 87
This job will run very fast, but if we needed to we could use commands like squeue and sinfo to examine the state of the cluster and jobs while it was running.
When the job is finished we can look at /shared_storage/mpi-ping-pong-87.err and /shared_storage/mpi-ping-pong-87.out to see all the output from our application. In this case, the error file is empty, but our output file looks like this:
Date start = Tue May 14 10:15:02 PM UTC 2024
Initiating Host = worker-0
Working Directory = /shared_storage
Number of Nodes Allocated = 3
Number of Tasks Allocated = 3
Hello, World! I am 🏓 Player (Rank) 1 . Process 2 of 3 on worker-1
Player (Rank) 1 's receiving 🏓 neighbor is Player (Rank) 0 and my sending 🏓 neighbor is Player (Rank) 2.
Hello, World! I am 🏓 Player (Rank) 2 . Process 3 of 3 on worker-2
Player (Rank) 2 's receiving 🏓 neighbor is Player (Rank) 1 and my sending 🏓 neighbor is Player (Rank) 0.
Hello, World! I am 🏓 Player (Rank) 0 . Process 1 of 3 on worker-0
Player (Rank) 0 's receiving 🏓 neighbor is Player (Rank) 2 and my sending 🏓 neighbor is Player (Rank) 1.
Player 0 sent the 🏓 ball to Player 1.
Player 1 received the 🏓 ball from Player 0.
Player 1 sent the 🏓 ball to Player 2.
Player 2 received the 🏓 ball from Player 1.
Player 2 sent the 🏓 ball to Player 0.
Player 0 received the 🏓 ball from Player 2.
Player 0 started round 2 by sending the 🏓 ball to Player 1.
…
Player 0 started round 900 by sending the 🏓 ball to Player 1.
🛑 Player 0 is bored of 🏓 ping pong and is quitting! 🛑
Player 1 received the 🏓 ball from Player 0.
Player 1 sent the 🏓 ball to Player 2.
🛑 Player 1 is bored of 🏓 ping pong and is quitting! 🛑
Player 2 received the 🏓 ball from Player 1.
🎉 Player 2 won the game of 🏓 ping pong, since everyone else quit! 🎉
Exit code = 0
Date end = Tue May 14 10:15:03 PM UTC 2024
If you look closely at the Hello, World! lines for each rank/process you will notice that they are all running on completely different nodes (worker-[0-2]), but the application still behaves exactly as we expect it to, and the messages are happily passed between each process running on a different core on different systems. Because of this, we could use Slurm to easily run our application across hundreds or even thousands of cores if that is what we needed.
Kubernetes
So, could we do all of this with Kubernetes today? Well, yes and no.
The real power of Slurm is that you have so much control over how it is built, how you assign resources to your workloads, and how they are partitioned while running.
MPI - Message Passing Interface
Although you could easily run this application inside a single pod on Kubernetes, to truly leverage the MPI capabilities that we are expecting we would need to install and utilize something like KubeFlow along with KubeFlow’s beta MPI Operator to even get started. You might also want to consider something like the Kubernetes-native Job Queueing support.
At the time of writing this KubeFlow v.1.8.0 doesn’t support versions of Kubernetes greater than v1.26.
One obvious side effect of using Kubernetes is that we MUST run our code inside one or more containers. This is possible with Slurm but is not a hard requirement. We will also need to ensure that Python, MPI, and sshd are all installed and configured correctly in our container, as we will need them all to run our application and manage each of the MPI worker pods. sshd is required because this is how mpirun will connect to each of the pods to launch the parallel instances of our application. The manifest that we will need to apply to our Kubernetes cluster for use with mpi-operator is simply named mpijob.yaml.
You can also find the Dockerfile for the container that we are running in the GitHub repo.
mpijob.yaml
apiVersion: kubeflow.org/v2beta1
kind: MPIJob
metadata:
name: mpi-ping-pong
spec:
slotsPerWorker: 1
sshAuthMountPath: /app/.ssh
runPolicy:
cleanPodPolicy: Running
ttlSecondsAfterFinished: 120
mpiReplicaSpecs:
Launcher:
replicas: 1
template:
spec:
containers:
- image: superorbital/mpi-ping-pong:latest
name: mpi-launcher
securityContext:
runAsUser: 5000
command:
- mpirun
args:
- -np
- "3"
- /app/mpi-ping-pong.py
resources:
limits:
cpu: 1
memory: 1Gi
Worker:
replicas: 3
template:
spec:
containers:
- image: superorbital/mpi-ping-pong:latest
name: mpi-worker
securityContext:
runAsUser: 5000
command:
- /usr/sbin/sshd
args:
- -De
- -f
- /app/.sshd_config
resources:
limits:
cpu: 1
memory: 1Gi
Assuming that Kubeflow and the mpi-operator are properly installed in our cluster, then applying the MPIJob manifest should “just work”.
$ git clone https://github.com/superorbital/mpi-playground.git
$ cd mpi-playground
$ kubectl apply -f ./manifests/mpi-ping-pong/mpijob.yaml
mpijob.kubeflow.org/mpi-ping-pong created
NOTE: If you want to try this out yourself, you will likely find that it is easiest to delete the MPIJob and then re-apply it each time you want to run it.
kubectl delete mpijobs.kubeflow.org mpi-ping-pong
In the animated GIF below you will see the manifest being launched, followed by the launcher and worker pods being spun up, once the workers are done, then the launcher will complete and the launcher logs will be displayed.
The sequence of events looks something like this:
- The required number of workers are started and
sshdis run inside each worker container. - The launcher is started and executes the desired
mpiruncommand. -
mpirunconnects to each worker and starts the individual tasks. - The workers complete their tasks and exit.
- The launcher completes and exits.
The pod logs for the launcher will contain all of the output that we would normally expect to see from an mpirun session.
So, today it is technically possible to run at least some MPI-style jobs inside Kubernetes, but how practical is it?
Well, one of the big advantages of Kubernetes is its ability to scale. Unlike Slurm, which has no cluster scaling functionality, a Kubernetes cluster can be designed to scale up nodes for a very large workflow and then scale back down once that workflow is finished and those nodes are no longer required for any immediately upcoming workflows.
But, I think the real answer here is that it depends a lot on what you are trying to do, how much control you have over the Kubernetes cluster setup, and how much effort you want to put into getting it all working.
Artificial Intelligence and Machine Learning
Although the technical details are different, most of the basic observations in this article also apply to artificial intelligence (AI11), machine learning (ML12), and deep learning (DL13) applications. In most instances, these workloads will make heavy use of systems equipped with advanced GPUs to significantly improve the performance of their computation tasks. Ensuring that each process has exclusive access to the required hardware resources is often critical for the success and performance of the task.
Slurm on Kubernetes
Over the last couple of years, Tim Wickberg, SchedMD’s CTO, has given a talk entitled “Slurm and/or/vs Kubernetes”, which looks at Slurm and Kubernetes and explores how each of these tools could be extended to leverage their strengths and generally improve the current state of high-performance workload managers.
One project that has come out of the effort to find synergy between these two tools and communities, is SUNK, which stands for SlUrm oN Kubernetes. It is a project in early development by SchedMD and CoreWeave to enable Slurm workloads in Kubernetes Clusters. The CoreWeave talk that officially introduced this project can viewed on YouTube if you would like to hear more details about the work that is being done with this. You can also grab a PDF of the slides for the talk. Once the code is released it should be available at github.com/coreweave/sunk.
Kubernetes Device Management
The Smarter Device Project from ARM research14 is also an interesting Kubernetes application that might be useful for these sorts of workloads and makes it possible to expose, reserve, and manage any Linux device (e.g. /dev/video*) within Kubernetes.
For GPUs, NVIDIA has a device plugin to provide similar functionality, which is often deployed along with their gpu-operator. This also enables features in Kubernetes like GPU time-slicing, something similar to the generic GPU sharding available in Slurm.
In Conclusion
Slurm is a very useful tool for running parallel applications that require very specific resources inside a compute cluster. It was designed for a set of workloads that are outside the standard stateless microservice focus of Kubernetes, but even today Kubernetes can be made to work with some of these workloads without too much hassle, however, truly leveraging the advantages of Kubernetes for these more traditional high-performance computing workloads will require projects like SUNK and mpi-operator to reach a reasonable level of maturity and then in the future, there may be newer projects that rethink the current approaches and build Kubernetes-first tools from the ground-up that leverage the strengths that are already available in the platform while extending its ability to handle the type of workloads that have been traditionally handled by tools like Slurm.
Further Reading
- Machine learning, explained
- Machine Learning Tutorial
- Introduction to MPI
- A Comprehensive MPI Tutorial Resource
- Managing Slurm at Scale
-
Intro to Multi-Node Machine Learning
- 1: Setting up an HPC cluster
- 2: Using Slurm
- Unreleased as of 05/16/2024 - 3: Multi-Node Training
- Introduction to Kubeflow MPI Operator and Industry Adoption
Acknowledgments
- Cover image by dlohner from Pixabay
- Slurm architecture overview by SchedMD for their network overview.